Question 1
Can you summarize three main genome browsers? Use them to explore human TERT gene and answer:
a) Its chromosome location and total length.
b) How many exons this gene has?
c) What’s the function of TERT protein? You can use other databases.
SCI4016-N Advanced Bioinformatics Assignment-Tees side University UK

Question 2
Consider a human disease for which a gene has been implicated (such as APP in Alzheimer’s disease) and a mouse model is available. How can forward genetics approaches can be used to study this disease? How can reverse genetics approaches can be used? What are some of the differences
in the kinds of information these two approaches can provide?
Question 3
The complete genome of bat corona virus Ra TG 13 has a NCBI accession number: MN996532, and the reference genome of SARS-Cov-2 has a NCBI assession number: NC_045512.2
a) Compare the length, number of genes, length of each gene of these two corona virus genomes.
b) Find the top 10 most similar SARS-cov2 genomes from human covid19 patients using NCBI BLAST for MN996532 and NC_045512 against beta corona virus database respectively, and compare the results, e.g., outcome genome length, identity. Need to specify the parameters used in the searching.
Question 4
You have two protein sequences:
Sequence 1: FKHMEDPLE and Sequence 2: FMDTPLNE
Complete the global pairwise alignment using Needleman-Wunsch approach by filling the following table.
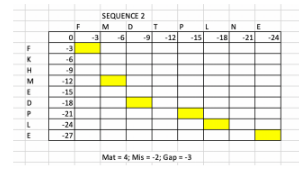
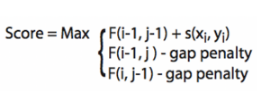
The formula above is used to infer the fourth value from three known value in a four-cell window. The alignment parameters: Match +4, Mismatch -2, Gap penalty -3
Your alignment result:
Sequence 1:
Sequence 2:
Question 5
GSE16515 data set is about the study of gene expression of pancreatic tumour using micro array technology. This dataset includes gene expression
from 36 pancreatic tumour samples and 16 normal control samples. Use GEO2R, excel and/or Network analyst to conduct the following analysis:
a) How many significant probesets (or genes) differently expressed between 36 tumour samples and 16 normal samples at the significant level of adj. pval < 0.05 (Benjamini & Hochberg) and log2FC > 1 (up-regulated) or log2FC < -1 (down-regulated)? We call these are DEGs. If we use raw p-value rather than adjusted p-value, i.e., P.value < 0.05 and log2FC > 1 or log2FC < -1, how many DEGs can you identify? We can conduct filtering in excel or programming in Python / R.
b) How many DEGs are up regulated and how many down regulated in these two DEGs lists respectively? You should give 4 numbers.
c) Make a volcano plot using excel or any other tools. We can use either adjusted or raw p-value.
d) Conduct KEGG pathway analysis for DEGs you identified. You can choose to use DAVID Pay attention to the type of IDs.
e) List the top 5 most significant DEGs identified by adjusted-p value approach in a), briefly interpret the most interesting gene you prefer to choose, e.g., its potential roles in pancreatic cancer, cite references if appropriate. Please do NOT paste the whole DEGs in your report document, only the top 5 DEGs.
Question 6
Please explain the whole data analysis process to conduct NGS for variant calling and complete the following NGS analysis using Galaxy. Need to indicate which galaxy tools you are going to use and specify the parameters.
Data: Human (pair-end) reads
Use the following file as your reference
a) How many reads included in the two fastq files and their read length respectively?
b) Conduct alignment using the provided reference genome and report the alignment statistics, e.g., apply sam tools flag stats. You can choose any aligner.
c) How many variants can you identify? You can download and open the VCF file in a text editor or excel and then count.
SCI4016-N Advanced Bioinformatics Assignment-Tees side University UK
Question 7
There are three GWAS cohort studies about SNP rs3923113, their OR, LOR, HOR (95%CI) and p-value in each study are:
Study 1: 1.15,1.09,1.21, 3.7×10-7
Study 2: 1.07,1.03,1.11, 6.7×10-4
Study 3: 1.05,1.01,1.10, 2.0×10-2
a) What’s the position of this SNP, its reference allele and nearest gene in human genome?
b) What is the combined p-value based on Fisher approach?
c) What is the combined p-value based on Fixed-effects model (Chi-square approach)?
d) What is the combined OR, LOR and HOR based on the combined beta and variance calculated in c)?
Question 8
What is machine learning and how can it be applied? What is the difference between supervised learning and unsupervised learning (give examples)? What is the cross-validation and for which purpose is it applied?
Question 9
In a classification problem, you have a confusion matrix as below for your classifier
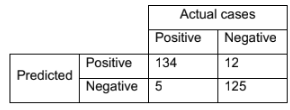
What is the accuracy, sensitivity and specificity values for this classifier in this problem? Can you explain what does ROC and AUC stand for?
Question 10
The compressed file ML_assess_2 Data sets.zip includes two normalized gene expression matrix files AD_train_small. arff (as training data set) and AD_test_small.aff (as testing data set) for patients with Alzheimer’s disease (AD) and health control (CTL) with a different number of samples. The columns represent the Gene IDs, followed by Class. Use the We ka software to answer the following questions:
a) How many AD/CTL samples are included in each dataset, and how many features (genes) are included?
b) For the training dataset, compare the classification performance of using SVM and logistic regression. Use 5-fold CV and indicate clearly the parameters, kernel you choose to use.
c) For the training dataset, apply feature selection approach to choose the top 10 features and then train your ML models (SVM and logistic regression). Thereafter, compare their performance with 5-fold CV.
d) Use the original AD_test_small.arff dataset to test SVM and logistic regression models trained by the original AD_train_small.arff dataset by applying “Supplied test set” rather than cross-validation. Compare the testing performance for these two ML models.
ORDER This SCI4016-N Advanced Bioinformatics Assignment NOW And Get Instant Discount

Read More :-

